StoJ and Modelling of Biochemical Pathways
In 2004, I was able to attend BioConcur ‘04. This led to some interesting thinking about alternative1 possibilities for modelling biochemical reactions, the meaning of bisimulation in process-based models of reactions, approaches to representing uncertainty in models, the choice of representation of state and concentration, and the need for a good model of reaction compartments.
A few weeks after the conference, these thoughts crystallised into a new join-calculus-based programming language, StoJ2, for exploring description, simulation, and visualization of biochemical systems modelled as concurrent, rate-limited processes.
The StoJ system
I’ve written a little about StoJ on my blog and on the LShift website:
-
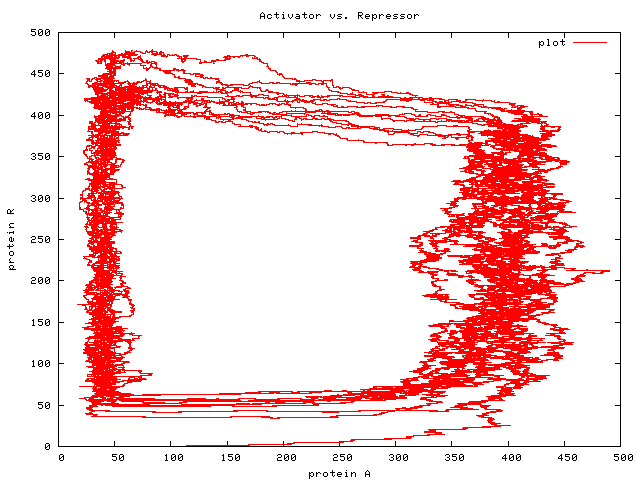
this article on the LShift website is a refinement of this earlier article, and not only sketches the language and its implementation, but describes the experiments we undertook with it to validate it against experiments described in the literature. (Happily, our experimental results lined up with those described in Regev01, showing that StoJ was a viable approach. The diagram above, from data produced by StoJ, agrees with a similar diagram in Regev01.)
-
this post fills in some of the background ideas we3 were thinking about on the road to building and experimenting with the StoJ system. It includes some discussion of bisimulation for biomolecular processes and so forth.
The language
The language is a polyadic, asynchronous stochastic π calculus with input join and no summation. Reaction rates are associated with a join instead of with a channel. The README describes the language syntax very briefly.
Here’s a simple chemical equilibrium model in StoJ:
// A simple H + Cl <--> HCl model,
// similar to that in Andrew Phillips' slides
// with numbers taken from his slides.
// See http://www.doc.ic.ac.uk/~anp/spim/Slides.pdf
// page 16.
new !h, !cl, !hcl . (
rec ASSOC. h() & cl() ->[100] (hcl<> | ASSOC)
| rec DISSOC. hcl() ->[10] (h<> | cl<> | DISSOC)
| h<> * 100 | cl<> * 100
)
Note the rates enclosed in square-brackets after the arrows on each join.
Simulations
See the main article for more complex examples and for information on using StoJ to simulate the behaviour of a particular process.
Visualizations
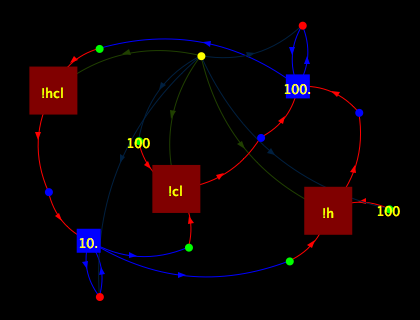
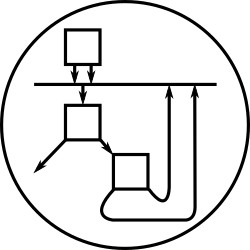
Besides charting the concentration-against-time data that the StoJ
simulator produces, the system includes a simple algorithm for
automatically transforming a process description into a a visual form
reminiscent of standard biochemical reaction schematics. The picture
on the left is a diagram produced automatically by StoJ from the HCl
model above; notice how the cycles in the diagram correspond to the
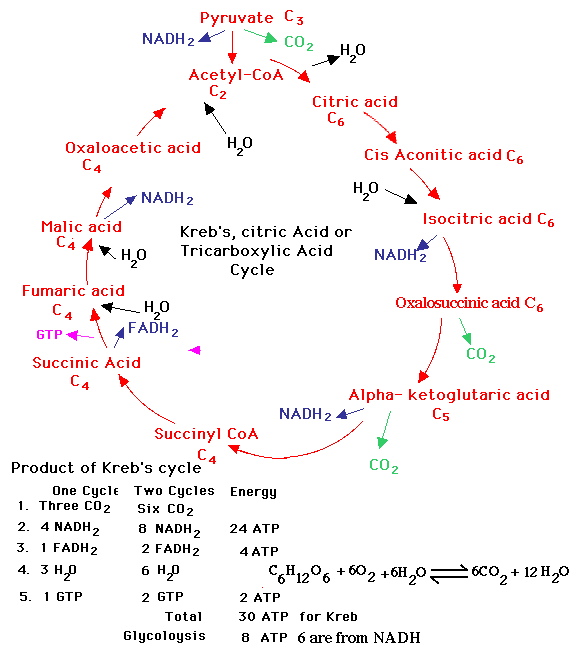
process’s reactions. Compare to the picture on the right, a standard,
hand-produced diagram of the Krebs cycle (taken from this
site).
Where the project went
StoJ hasn’t been developed any further since that initial burst of activity. It’s possible that using a join-calculus approach to biomolecular modelling is an idea worth following up on, but I haven’t had a chance to revisit it.
The broader project that provided a space for us to engage in this kind of thinking ended a few months after StoJ was produced, helping provide a foundation for the bioinformatics startup that commissioned LShift to help research pi-calculus-based modeling of biochemical pathways and investigate logics for describing whole classes of biochemical pathway.
Download
The code is a CGI-driven application in an unholy combination of OCaml, Python, and Java. Stoj is currently hosted on git.leastfixedpoint.com:
- download a current snapshot tarball, containing not just the source code, but also a little bit of documentation for the system
- browse the code
git clone https://git.leastfixedpoint.com/tonyg/stoj
References
Priami C, Regev A, Shapiro E, Silverman W. Application of a stochastic name-passing calculus to representation and simulation of molecular processes. Information Processing Letters. 2001;80(1):25-31. Available at: http://linkinghub.elsevier.com/retrieve/pii/S0020019001002149.
-
an alternative to the approach taken by the stochastic pi calculus of Priami, Regev, Shapiro and Silverman; see the BioSPI webpage for details. ↩
-
For Stochastic Join calculus. Never let computer scientists name things! ↩
-
mostly me and my colleague Michael Bridgen ↩